Workflows
What is a Workflow?Filters

Snakemake workflow: FAIR CRCC - image conversion
![]()
![]()
A Snakemake workflow for converting whole-slide images (WSI) from the CRC Cohort ...
Snakemake workflow: dna-seq-varlociraptor
![]()
![]()
![]()
A ...
A prototype implementation of the Air Quality Prediction pipeline in Galaxy, using CWL tools.

ABR_Threshold_Detection
What is this?
This code can be used to automatically determine hearing thresholds from ABR hearing curves.
One of the following methods can be used for this purpose:
- neural network (NN) training,
- calibration of a self-supervised sound level regression (SLR) method
on given data sets with manually determined hearing thresholds.
Installation:
Run inside the src directory:
Installation as python package
pip install -e ./src (Installation as python
...


StructuralVariants Workflow
Type: Common Workflow Language
Creators: Laura Rodriguez-Navas, Daniel López-López
Submitter: Laura Rodriguez-Navas
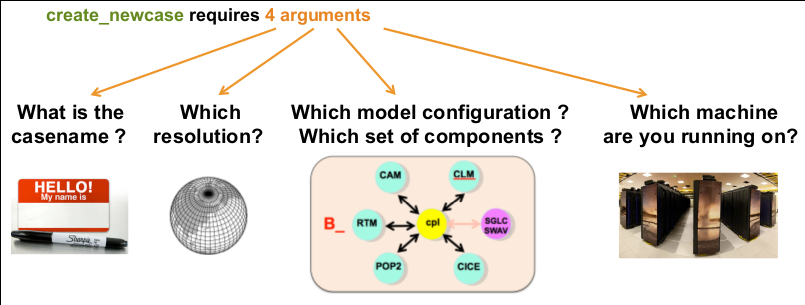
This workflow demonstrates the usage of the Community Earth System Model on Galaxy Europe.
A fully coupled B1850 compset with resolution f19_g17 is run for 1 month.

HiFi de novo genome assembly workflow
HiFi-assembly-workflow is a bioinformatics pipeline that can be used to analyse Pacbio CCS reads for de novo genome assembly using PacBio Circular Consensus Sequencing (CCS) reads. This workflow is implemented in Nextflow and has 3 major sections.
Please refer to the following documentation for detailed description of each workflow section:
Generic consensus building
This workflow generates consensus sequences using a list of variants generated by Variant Calling Workflow.
The workflow accepts a single input:
- A collection of VCF files
The workflow produces a single output:
- Consensus sequence for each input VCF file
The workflow can be accessed at usegalaxy.org
Generic variation analysis reporting
This workflow generates reports from a list of variants generated by Variant Calling Workflow.
The workflow accepts a single input:
- A collection of VCF files
The workflow produces two outputs (format description below):
- A list of variants grouped by Sample
- A list of variants grouped by Variant
Here is example of output by sample. In this table all varinats in all samples are epxlicitrly listed:
| Sample | ...
Generic variant calling
A generic workflow for identification of variants in a haploid genome such as genomes of bacteria or viruses. It can be readily used on MonkeyPox. The workflow accepts two inputs:
- A genbank file with the reference genomes
- A collection of paired fastqsanger files
The workflow outputs a collection of VCF files for each sample (each fastq pair). These VCF files serve as input to the Reporting workflow.
Workflow can be accessed ...