Workflows
What is a Workflow?Filters
Just the cleaning then assembly of all reads. TO explore further follow one of the paths described in "Global view" (WF 0)
 Download
DownloadMapping against all plant virus then make contig out of the mapped reads then blast them.
extract 1 Id from SRA and assume it is PE as input to viralRNASpades.





- Deprecated -
See our updated hybrid assembly workflow: https://workflowhub.eu/workflows/367
And other workflows: https://workflowhub.eu/projects/16#workflows
Workflow for sequencing with ONT Nanopore data, from basecalled reads to (meta)assembly and binning
- Workflow Nanopore Quality
- Kraken2 taxonomic classification of FASTQ reads
- Flye (de-novo assembly)
- Medaka (assembly polishing)
- metaQUAST (assembly quality reports)
When Illumina reads are provided:
- Workflow ...
Type: Common Workflow Language
Creators: Bart Nijsse, Jasper Koehorst, Germán Royval
Submitter: Jasper Koehorst
1. About TF-Prioritizer
This pipeline gives you a full analysis of nfcore chromatine accessibility peak data (ChIP-Seq, ATAC-Seq or DNAse-Seq) and nfcore RNA-seq count data. It performs DESeq2, TEPIC and DYNAMITE including all preprocessing and postprocessing steps necessary to transform the data. It also gives you plots for deep analysis of the data. The general workflow is sketched in the images below:
Graphical abstract:
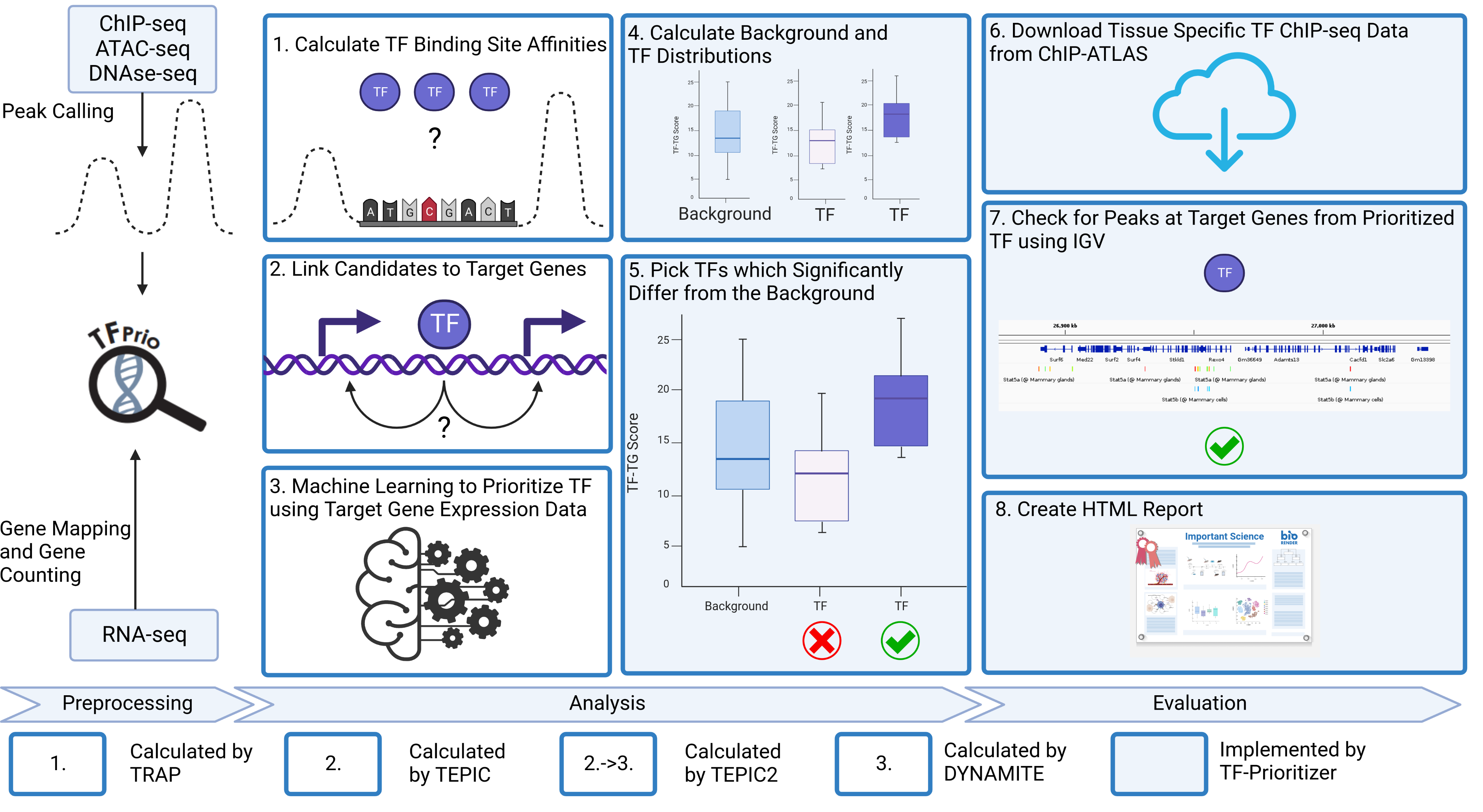 ...
...

Snakemake workflow: Reconstructing raw tomography data
A Snakemake worfklow for tomographically reconstructing raw data using tomopy.
Installation
First download this repo and navigate to it
git clone https://codebase.helmholtz.cloud/gernha62/reconstructing-raw-tomography-data.git
cd /path/to/repo
(Optional) Download the example folder with:
wget -m -np https://doi2.psi.ch/datasets/das/work/p15/p15869/compression/MI04_02/tif
...

TronFlow BAM preprocessing pipeline

![]()
![]() ...
...
TronFlow alignment pipeline

![]()
![]() ...
...
![]()
CoVigator pipeline: variant detection pipeline for Sars-CoV-2
![]()
![]() [![Powered by
...
[![Powered by
...
Type: Nextflow
Creators: Pablo Riesgo Ferreiro, Thomas Bukur, Patrick Sorn
Submitter: Pablo Riesgo Ferreiro
Github: https://github.com/Lcornet/GENERA
BCCM GEN-ERA tools repository
Please visit the wiki for tutorials and access to the tools: https://github.com/Lcornet/GENERA/wiki
NEWS
Mantis is now installed in a singularity container for the Metabolic workflow (install is no longer necessary).
Information about the GEN-ERA project
Please visit https://bccm.belspo.be/content/bccm-collections-genomic-era
Publications
- ToRQuEMaDA: tool for retrieving queried Eubacteria, metadata and dereplicating ...